Orthology and paralogy
When describing the evolutionary past of a gene, we call those genes that have common descent homologs. A more detailed description of a pair of genes can be done using terms orthologs for those homologs that were derived in the event of speciation and paralogs for those homologs that are separated by an event of duplication.
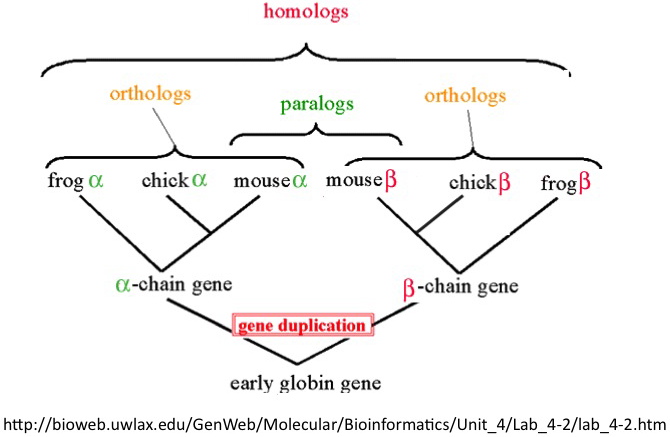
As these terms are defined in light of the phylogenetic tree, the strict definition demands we have the phylogenetic relations between organisms resolved prior to the derivation of orthologous and paralogous relationships. In the strict sense, these relations can be described for pairs of genes only. Due to the fact that the construction of phylogeetic trees is far from being trivial, there are numerous methods that are based on the use of sequence homology (without relying on the phylogenetic tree) to define the orthologous and paralogous relationships. Given the fact that we focus on prokaryotic organisms that do not have fully resolved phylogenetic relationships (owing to a large amount of horizontal gene transfer), we turn our attention to these methods. Also, the grouping of pairs of orthologs will be useful when we construct our multi-genome datasets.
OMA database
The OMA algorithm is a sequence homology-based method of orthology inference that accounts for events of gene loss: bidirectional strong hits (bidirectional best hits, within a confidence interval) from two species are broken when a third species (“witness to non-orthology”) can be found, containing a pair of proteins more similar to the respective proteins in an evaluated pair than the bidirectional strong hit proteins are similar to each other. The pairs for which a witness to non-orthology was shown to exist are termed “broken pairs” i.e., paralogs; the remaining pairs (true orthologs) are used to form OMA groups. It has a large coverage of the sequenced organisms, with regular updates and can be found at http://omabrowser.org/.
© 2010 Design by: styleshout Webmaster: mb
Contact:
GORBI: Gene Ontology at Ruđer Bošković Institute by http://gorbi.irb.hr is licensed under a Creative Commons Attribution-Non-Commercial-Share Alike 3.0 Croatia License.