OMA orthologs and paralogs
We based our functional annotation models on orthologous and paralogous pairs of proteins inferred by the OMA algorithm (Roth et al, 2008, Altenhoff et al, 2011), briefly explained below. In addition to being available as a standalone software, the OMA results are available for browsing online.
OMA is a graph-based method of orthology inference. The algorithm starts with an all-against-all sequence alignment: proteins from two species are connected if they are best bidirectional hits, allowing for a confidence interval.
Next, these connections may be broken if a third species contains a pair of proteins more similar to each of these two proteins than the connected proteins are similar to each other. Such broken pairs are inferred paralogs, while the remaining connections are inferred orthologs. Finally, OMA ortholog cliques are sub-graphs where all proteins are connected by orthology relationships.
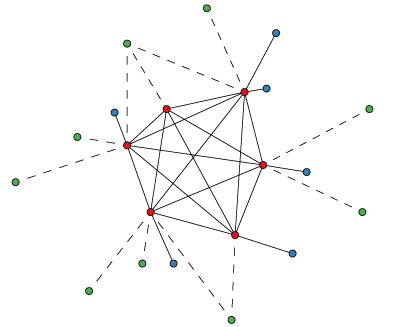
Based on the above, we constructed four kinds of phylogenetic profiles:
- First, phylogenetic profiles of OMA cliques of orthologs: each profile represents the pattern of presence/absence of an OMA clique member among 909 Bacterial and 89 Archaeal genomes.
- Second, we added the presence patterns for all remaining orthologs inferred by the OMA algorithm that did not participate in the original ortholog clique.
- Third, we added presence patterns for all paralogs inferred by the OMA algorithm.
- Fourth, we also made a separate set of phylogenetic profiles that include only clique members and paralogs, but not the orthologs outside of the clique.
© 2010 Design by: styleshout Webmaster: mb
Contact:
GORBI: Gene Ontology at Ruđer Bošković Institute by http://gorbi.irb.hr is licensed under a Creative Commons Attribution-Non-Commercial-Share Alike 3.0 Croatia License.