Hierarchical multi-label classification
In order to predict GO annotations of OMA orthologous groups, we employed the machine learning algorithm CLUS-HMC, based on decision trees for Hierarchical Multi-label Classification. The CLUS-HMC decision trees are able to deal with multiple, hierarchically organized class labels. In our case, these are GO annotations of genes.
Clus-HMC can be extended to an ensemble (CLUS-HMC-Ens); in particular, we use a Random Forest - a large number of decision trees constructed from bootstrap samples of the data. An implementation of the Clus-HMC is available.
We compared the predictive performance of the CLUS-HMC Random Forest to a standard approach in analysis of gene functional interactions.
In particular, this means inferring the function of a gene by looking at its closest nodes in a network - here, the genes with most similar phylogenetic profiles. This is known as the k-nearest neighbors algorithm. The CLUS-HMC random forest considerably outperforms this baseline approach (see below).
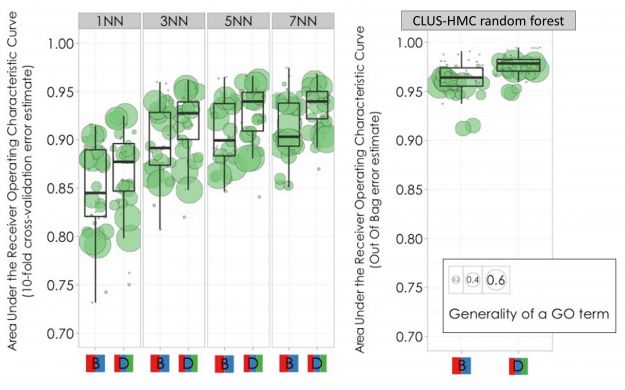
Figure S9 from the GORBI paper. Comparing the CLUS-HMC Random Forest with the k-nearest neighbors (kNN) classifiers in predicting GO functions from the Cellular Component ontology. The panels marked "B" contain phylogenetic profiles of orthologs, while "D" denotes orthologs+paralogs. The y-axis is the crossvalidation accuracy as Area Under the Precision-Recall Curve (AUPRC).
© 2010 Design by: styleshout Webmaster: mb
Contact:
GORBI: Gene Ontology at Ruđer Bošković Institute by http://gorbi.irb.hr is licensed under a Creative Commons Attribution-Non-Commercial-Share Alike 3.0 Croatia License.